Sickle cell disease therapeutic target identified
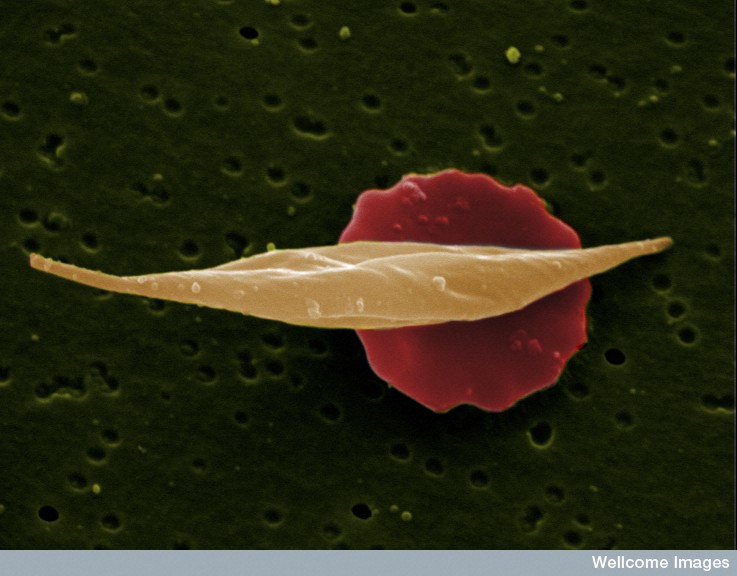
Caption: A sickle-shaped blood cell in front of an unaffected red blood cell. EM Unit, UCL Medical School, Royal Free Campus, Creative Commons license: CC BY-NC-ND 2.0

When Elenoe “Crew” Smith joined Stuart Orkin’s lab at Boston Children’s Hospital, the group had just identified a segment of DNA that could prove pivotal for sickle cell disease research.
A critical gene, BCL11A, determines whether or not a red blood cell will produce the adult form of hemoglobin. In people with sickle cell disease, the adult form of hemoglobin is mutated, creating misshapen red blood cells that lead to anemia, stroke, multiple organ failure, and pain. The fetal form is unaffected by the disease. The DNA segment in question, called an enhancer, promoted the conversion of fetal hemoglobin to adult, and –if it only functioned in red blood cells– it had the potential to be a valuable therapeutic target.
“To analyze and confirm the red blood cell specificity of this element, I chose to engineer a mouse lacking the enhancer in every cell of its body,” says Smith, a JCC Fellow. “This was particularly challenging since we were trying to remove a rather large segment of DNA from the mouse genome.” She spent a year and a half using conventional homologous recombination before trying a different technology known as TALEN-based genome editing. Neither worked. “Then I switched to the CRISPR/Cas9 system, which allowed me to isolate embryonic stem cell clones within a month and a half,” she says. “I gave our mouse transgenic core facility the modified mouse embryonic stem cells right before I went on maternity leave. By the time I got back, I had mice ready to be analyzed.”
Smith and her colleagues found that indeed the enhancer was specific to red blood cells. Further experiments show that cuts made to a specific region of the enhancer resulted in higher levels of the benign fetal hemoglobin. The work, published in Nature, was a collaboration among the labs of Orkin, Daniel Bauer also of Boston Children’s Hospital, and Feng Zhang of the Broad Institute of MIT and Harvard.
Smith’s path to becoming a JCC Fellow began with her upbringing in the United States Virgin Islands. Her parents worked at the university there, her mother as an accountant and her father as a principal investigator on water catchment and disposal research funded by the National Science Foundation. “My family valued learning and I was expected to pursue higher education,” she says.
After her parents saw a PBS special about a unique financial aid process at Princeton that offered grants instead of loans, Smith applied and was accepted as a molecular biology major. “My first semester was very challenging, I was among students with not only exceptional academic talent but also world class musicians and athletes. I was no longer a big fish.”
Smith recalls that by her senior year, she had two amazing mentors who encouraged her to attend graduate school at an institution that supported minorities and women. She worked toward her PhD with Professor Diane Krause in the Cell Biology Department at Yale University.
While at Yale, Smith co-organized symposia highlighting minority faculty at Yale. She has also returned to the Virgin Islands to share her experiences in science and higher education with high schoolers. “Undergraduate and graduate school mentors were instrumental in my success and I try to provide that support to other underrepresented minorities in science,” Smith says.